
Herediter Spastik Paraplejiler
Giriş
Herediter spastik paraplejiler bacaklarda kas güçsüzlüğü ve spastisite (sertlik) ile seyreden gen mutasyonları (bozuklukları) nedeniyle oluşan bir hastalık grubudur. Herediter kalıtsal, spastik anormal kas kasılması ve parapleji de bacaklarda kas güçsüzlüğü demektir. Hastalığın sıklığı 100.000 kişide 1 ile 10 arasında değişmektedir.
Herediter spastik paraplejiler tek bir hastalık değil, bir hastalık grubudur. Çünkü bugüne kadar 70’den fazla gen mutasyonunun hastalığa sebep olduğu gösterilmiştir. Mutasyon bir genin görevini yapmasını engelleyen bir değişiklik halidir. Mutasyon olduğunda gen olması gerektiği gibi çalışamaz ve vücuttaki görevini yeterince gerçekleştiremez. Bazı sinir hücrelerinin akson dediğimiz uzantıları beyinden omuriliğe kadar 1 metre boyunca devam etmektedir. Bu kadar uzun bir hücrenin son kısmının, başlangıç kısmında yer alan gövdeden yeterince beslenebilmesi, enerji ihtiyacını karşılayabilmesi için çok sayıda genin görev yapması gereklidir. Herediter spastik paraplejilere 70’den fazla genin mutasyonun neden olmasının sebebi budur. Bu gen mutasyonu sinir hücrelerinin yapısını, beslenmesini, hücreler arası iletişimi bozarak hastalık belirtilerinin ortaya çıkmasına neden olur. Şekil 1’de şematik olarak herediter spastik paraplejilerin oluş mekanizmaları anlatılmıştır.
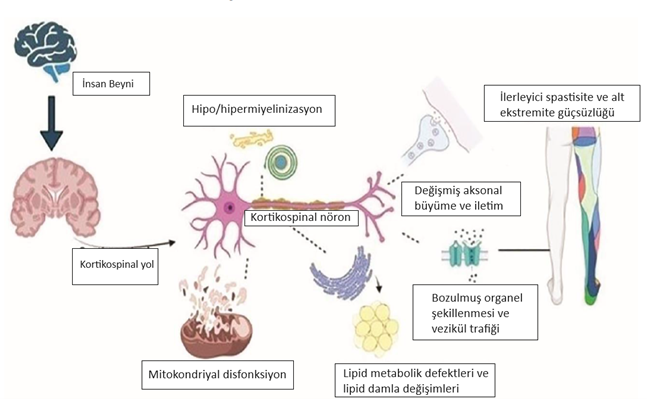
Şekil 1. Herediter spastik paraplejilerde biyolojik fonksiyon bozukluğu ve nedenleri. Beynin dış kabuğundan omuriliğe kadar uzanan sinir hücrelerinde oluşan mutasyonlar mitokondri, vezikül, miyelin gibi yapıtaşlarının görevlerini yapamamasına neden olur. Sinir hücresinin metabolizmasının bozulması ile bacaklarda güçsüzlük ve spastisite belirtileri ortaya çıkar (1).
Genetik sınıflandırma
En sık görülen çeşidi hastaların %75-80 kadarını oluşturan otozomal dominant kalıtımlı olandır. DNA’mızda her genin biri anneden biri babadan geçen iki kopyası bulunmaktadır. Otozomal dominant kalıtımlı demek bir gendeki iki kopyadan sadece birinde mutasyon olmasıdır. SPAST ve ATL1 genlerinin mutasyonu bu gruptaki olguların neredeyse yarısından sorumludur. Bu tipte herediter spastik paraplejili bir bireyin çocuklarına hastalığı %50 oranında geçirme ihtimali vardır. Anne, baba, kardeş gibi başka bir aile bireyinde hastalık olmadan da ilk kez o kişide otozomal dominant herediter spastik parapleji oluşabilir.
Genellikle akraba evliliği ile ilişkili olan otozomal resesif grup ise tüm hastaların %25-30 kadarını oluşturur. Otozomal resesif kalıtımlı demek ise bir gendeki iki kopyanın da mutasyona uğramış olmasıdır. Bu grupta hastalık nedeni olabilecek gen sayısı çok fazladır. Bazı genlerin mutasyonu ile oluşan hastalık o kadar nadirdir ki dünya üzerinde sadece bir veya birkaç kişide bulunabilir. CYP7B1, SPG7, SPG11 genleri bu gruptadır. Bu tipte ise hastanın anne ve babası hastalığın taşıyıcısıdır. Hastalığın %25 oranında kardeşlerde tekrarlama riski bulunmaktadır. X kromozomu ve mitokondri ilişkili kalıtımla olanlar ise %1-2 kadardır.
Tablo 1. Hastalık tipi, kalıtım modeli ve etkilenen gen ile tüm herediter spastik parapleji tipleri sunulmuştur. (2)
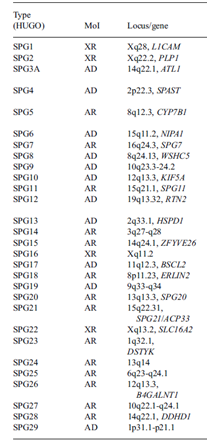
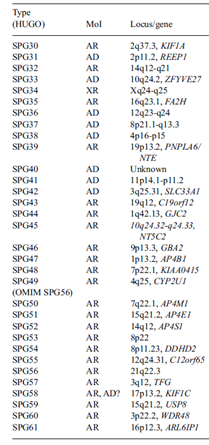

Type:Tip, SPG:spastic paraplegia (spastik parapleji), AR:autosomal recessive (otozomal resesif), AD:autosomal dominant (otozomal dominant), XR: X-linked recessive (X’e bağlı resesif), Gene: gen, Locus:Lokus, genin kromozomdaki yeri
Belirtilere göre sınıflandırma
Hastalık belirtileri temelde iki şekilde seyretmektedir; saf (komplike olmayan) ve komplike. Saf tipte belirtiler sadece hastalığın isminde olduğu gibidir yani bacaklarda yavaş ilerleyen güçsüzlük ve spastisite (anormal kas kasılması) mevcuttur. Bazı hastalarda idrar kontrolünde güçlükler de olabilir. Saf tipte belirtiler sağlıklı normal bir dönemden sonra genellikle erişkin yaşta başlar. Belirtilerde yavaş bir hızda artış görülür. Komplike tipinde ise hastalığın temel belirtileri olan bacaklarda güçsüzlük ve anormal kas kasılmasına başka nörolojik belirtiler de eşlik eder, örneğin dengesiz yürüme, ellerde titreme, zihinsel kısıtlılıklar, epilepsi, nöbet, distoni (kıvrılma tarzı kas kasılmaları), nöropati (kol/bacak sinirlerinde etkilenme). Bu belirtilerden bir veya birkaçı bir arada bulunabilir. Komplike tipte belirtiler genellikle çocukluk çağında başlamaktadır.
Hastalığın hangi belirtilerle seyredeceğini, ne zaman başlayacağını, hangi hızda ilerleyeceğini belirleyen altta yatan genetik neden yani etkilenen gen ve mutasyondur. Başlangıç yenidoğandan ileri yaştaki yetişkinlere kadar geniş bir aralıkta olabilir. Ancak aynı ailede aynı gen ve mutasyon olmasına rağmen bir birey hafif, diğeri ağır etkilenmiş olabilmektedir. Burada mutasyon olan gen dışındaki diğer genlerin olumlu veya olumsuz etkisi söz konusudur.
Tanı testleri
Tanı aşamasında hastanın ailesindeki benzer bireyler sorulmalıdır. Detaylı öykü (belirtilerin başlama zamanı, ilerleme hızı, diğer belirtilerin varlığı), nörolojik muayene ile birlikte ayrıntılı fizik muayene önemlidir. Herediter spastik paraplejiler serebral palsi başta olmak üzere benzer belirtilere neden olabilecek hastalıklarla karışabilir. Öykü, fizik muayene bulgularına göre uygun tetkikler (örn: beyin ve omurilik MRG) planlanabilir.
Genetik testler
Belirti ve bulgularla herediter spastik paraplejiden şüphelenilen hastalarda kesin tanı mutasyona uğramış olan genin genetik testlerle saptanmasıdır. Genetik testler bir tüp kan alınarak yapılmaktadır. Kesin tanının herediter spastik paraplejiye neden olduğu bilinen genleri içeren genetik paneller (az sayıda gen incelenir), klinik ekzom dizileme (yaklaşık 5000 civarında geni analiz eder) veya tüm ekzom dizileme (22000 civarında geni analiz eder) incelemelerinden biriyle konulması mümkün olabilir. Örneğin eğer sadece panel yapılan bir hastada tanı bulunamadıysa daha çok geni test eden incelemelerden yardım almak gerekir. Her geçen sene daha önce bilinmeyen yeni genlerin hastalığa neden olduğu bulunmaktadır. Bir genetik neden bulunamaması hastalığın olmadığı anlamına gelmemektedir. İncelenen gen sayısını arttıracak şekilde testleri sürdürmek gerekebilir.
Tedavi ve takip
Tedavide ne yazık ki henüz belirtileri tamamen ortadan kaldıracak bir ilaç yoktur. Ancak genetik kökenli hastalıklarda son yıllarda büyük ilerlemeler olmaktadır. Ümidimiz gelecekte hastalığa neden olan başta en sık genler olmak üzere tüm genler için bir tedavi bulunmasıdır. Tam iyileşme sağlayacak tedavi olmamasına rağmen semptomları azaltmak için yapılabilecekler mevcuttur. Tedavinin amacı hareket kapasitesini iyileştirmek, hareket açıklığını arttırmak, spastisiteyle ilişkili rahatsızlığı azaltmaktır. Tedavi ilaçlar ve fizik tedavi/rehabilitasyon olarak iki gruba ayrılır. İlaç tedavisi spastisite dediğimiz anormal kas kasılmasını iyileştirmek için kullanılanlardır. Ağızdan baklofen, tizanidin ve kaslara enjeksiyon şeklinde uygulanan botulinum toksinidir.
Önemli bir diğer nokta, hastalara veya hastaların ebeveynlerine genetik danışmanlık sağlamaktır. Böylelikle hastalığın kardeşlerde veya hastanın kendi çocuklarında tekrarlama riski belirlenebilir. Bazı durumlarda anne karnındaki bebeğe de genetik test yapmak mümkün olabilir.
Seyir
Saf (komplike olmayan) herediter spastik paraplejiler yaşam süresi üzerini kısaltmazlar ancak hayat kalitesini etkileyebilirler. Belirtilerin başlangıç yaşı ortalamasının 30 yaş olduğu bir araştırmada hastalığın 22 yılı (medyan) boyunca hastalar bağımsız yürümeye devam edebilmiştir. Tekerlekli sandalye kullanımı 37 yıllık (medyan) hastalık süresinin sonunda gerçekleşmiştir.
Kaynaklar:
- Meyyazhagan A, Orlacchio A. Hereditary Spastic Paraplegia: An Update. Int J Mol Sci. 2022 Feb 1;23(3):1697. doi: 10.3390/ijms23031697.
- Blackstone C. Hereditary spastic paraplegia. Handb Clin Neurol. 2018;148:633-652. doi: 10.1016/B978-0-444-64076-5.00041-7.
- Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol. 2014 Jul;34(3):293-305. doi: 10.1055/s-0034-1386767.
- Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012 Jul 15;318(1-2):1-18. doi: 10.1016/j.jns.2012.03.025.
- Ayaz A, Uzunhan TA, Aydin K. Interacting with AP1 complex mutated synergin gamma (SYNRG) reveals a novel coatopathy in the form of complicated hereditary spastic paraplegia. Brain Dev. 2022 May;44(5):329-335. doi: 10.1016/j.braindev.2022.01.002.
- Topaloğlu H, Pinarli G, Erdem H, Gücüyener K, Karaduman A, Topçu M, Akarsu AN, Ozgüç M. Clinical observations in autosomal recessive spastic paraplegia in childhood and further evidence for genetic heterogeneity. Neuropediatrics. 1998 Aug;29(4):189-94. doi: 10.1055/s-2007-973559.
- Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019 Dec;18(12):1136-1146. doi: 10.1016/S1474-4422(19)30235-2.
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008 Dec;7(12):1127-38. doi: 10.1016/S1474-4422(08)70258-8.
- Schiavoni S, Spagnoli C, Rizzi S, Salerno GG, Frattini D, Pisani F, Fusco C. Paediatric-onset hereditary spastic paraplegias: a retrospective cohort study. Dev Med Child Neurol. 2020 Sep;62(9):1068-1074. doi: 10.1111/dmcn.14547.
- Schüle R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, Klimpe S, Gallenmüller C, Kurzwelly D, Henkel D, Rimmele F, Stolze H, Kohl Z, Kassubek J, Klockgether T, Vielhaber S, Kamm C, Klopstock T, Bauer P, Züchner S, Liepelt-Scarfone I, Schöls L. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann Neurol. 2016 Apr;79(4):646-58. doi: 10.1002/ana.24611.
Doç. Dr. Tuğçe Aksu Uzunhan
Çocuk Nöroloji Uzmanı
Çocukluk Çağı Nörodejeneratif Hastalıkları Araştırma Derneği