Nöronal Seroid Lipofuskinozis Tip 2 (NCL 2) Erken Tanı ve Tarama Çalışması
Araştırmanın Amacı
Çocukluk Çağı Nörodejeneratif Hastalıkları Araştırma Derneği tarafından yürütülecek bu çalışmada NCL Tip 2 hastalarının erken teşhisinin sağlanması amacıyla TPP1 enzim seviyesi testi yapılması tasarlanmıştır.
Araştırma yöntemi
Bir yıl boyunca çalışmanın yapılacağı merkezlere (Türkiye genelinde 35 merkez) 18 aylık ile 4 yaş arası olan ve asemptomatik nöbet ile hastaneye başvuran yanı sıra dil, bilişsel ve motor gelişim basamaklarında gerilik ya da gerileme bulgularından en azından birinin olduğu çocuklarda rutin alınan kan tetkikleri sırasında Guthrie kağıdına damlatılan birkaç damla kan ile TTP1 enzim çalışması yapılacaktır. Araştırma Sonunda Beklenen Yararlar Etkin bir enzim tedavisi bulunan NCL2 hastalığı olan çocuklarda erken teşhis ile tedaviye erken başlanmasının hastalık seyrinde oldukça etkili olması nedeniyle, bu hastalığı olan çocuklarda daha erken dönemde tedaviye ulaşım sağlanacaktır.
Biyotin Tiyamin Yanıtlı Bazal Gangliya Hastalığı Nedir?
Biyotin Tiyamin Yanıtlı Bazal gangliya Hastalığı (BTBGH), genetik (ırsi) kökenli nadir bir hastalıktır. Hastalık adını tedavisinde kullanılan vitaminlerden almaktadır. Bazal gangliyalar beynin iç kısmında bulunan hareketin kontrol edilmesinde, öğrenme gibi bilişsel işlevler, duygu ve düşüncelerin kontrolünde görev alır. Hastalığın görülme yaşı değişken olup çocukluk, erken bebeklik veya yetişkinlik döneminde ortaya çıkabilir.
BTBGH’ye ne sebep olur?
Anne babadan geçen bir gende meydana gelen mutasyon (bozulma) sonucu oluşan nörometabolik bir hastalıktır. Nörometabolik hastalıklar, vücudun kimyasal bir reaksiyon için ihtiyaç duyduğu besin, vitamin ya da enzimin olmamasından kaynaklanan, genetik kökenli, çoğunlukla ilerleyici bir grup hastalıktır. Anne baba akrabalığı olması durumunda görülme sıklığı artar ancak kendiliğinden ortaya çıkan genetik mutasyonlar sonucunda da oluşabilir. SLC19A3 geni, tiyamini hücrelere taşıyan bir proteinin üretim emrini verir. B1 vitamini olarak da bilinen tiyamin, günlük besinlerden elde edilir ve sinir sisteminin düzgün çalışması için gereklidir. Gendeki mutasyonlar tiyamini hücrelere taşıma kabiliyetine sahip olmayan bir proteine yol açar, bu da vitamin emiliminin azalmasına ve nörolojik işlev bozukluğuna yol açar.
Canavan hastalığı (CH) bebeklik döneminde başlayan beyin işlevlerinde gerilemeye yol açan ilerleyici bir nörolojik hastalıktır. Kalıtsal bir genetik bozukluktan kaynaklanır. Bu genetik bozukluk nedeniyle vücutta bulunan bir enzimin (aspartoasilaz: ASPA) eksikliği ortaya çıkar. Bu enzimin eksikliği beyindeki beyaz cevherin (miyelin) bozulmasına neden olur ve böylece sinir sinyallerinin iletimi yavaşlar.
CH’de hangi belirtiler görülür?
Canavan hastalığının belirtileri değişkenlik gösterir. Genellikle hızla büyüyen baş çevresi, baş tutmada zorluk, görmede azalma ve kol ve bacaklarda sertlik veya gevşeklik görülür. Bebeklik döneminde belirti veren Canavan hastalığı olan çocuklar emekleyemez, yürüyemez, oturamaz ve konuşamaz. Zamanla nöbet geçirebilir, gelişimsel olarak gecikebilir ve yutma güçlüğü çekebilirler. İşitme kaybı ortaya çıkabilir.
Belirtiler genellikle bebek üç ila dokuz aylıkken ortaya çıkar. Şu anda Canavan hastalığının tedavisi yoktur. Tedavi belirti ve bulgulara yöneliktir.
Herediter spastik paraplejiler bacaklarda kas güçsüzlüğü ve spastisite (sertlik) ile seyreden gen mutasyonları (bozuklukları) nedeniyle oluşan bir hastalık grubudur. Herediter kalıtsal, spastik anormal kas kasılması ve parapleji de bacaklarda kas güçsüzlüğü demektir. Hastalığın sıklığı 100.000 kişide 1 ile 10 arasında değişmektedir.
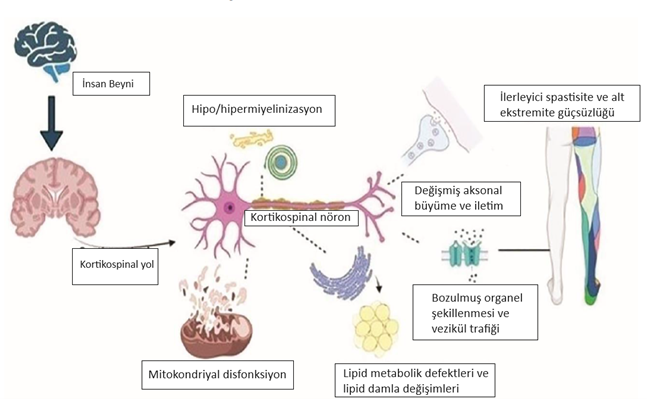
Herediter spastik paraplejiler tek bir hastalık değil, bir hastalık grubudur. Çünkü bugüne kadar 70’den fazla gen mutasyonunun hastalığa sebep olduğu gösterilmiştir. Mutasyon bir genin görevini yapmasını engelleyen bir değişiklik halidir. Mutasyon olduğunda gen olması gerektiği gibi çalışamaz ve vücuttaki görevini yeterince gerçekleştiremez. Bazı sinir hücrelerinin akson dediğimiz uzantıları beyinden omuriliğe kadar 1 metre boyunca devam etmektedir. Bu kadar uzun bir hücrenin son kısmının, başlangıç kısmında yer alan gövdeden yeterince beslenebilmesi, enerji ihtiyacını karşılayabilmesi için çok sayıda genin görev yapması gereklidir. Herediter spastik paraplejilere 70’den fazla genin mutasyonun neden olmasının sebebi budur. Bu gen mutasyonu sinir hücrelerinin yapısını, beslenmesini, hücreler arası iletişimi bozarak hastalık belirtilerinin ortaya çıkmasına neden olur. Şekil 1’de şematik olarak herediter spastik paraplejilerin oluş mekanizmaları anlatılmıştır.
Krabbe hastalığı, globoid hücreli lökodistrofi olarak da bilinen nadir bir genetik hastalıktır. Krabbe hastalığı, sinir hücrelerini çevreleyen koruyucu örtünün (miyelin kılıfı) kaybından kaynaklanan ciddi bir nörolojik durumdur. Bu koruyucu miyelin kılıfı, sinirleri yalıtmak ve sinir sinyallerinin vücutta hızlı iletimini sağlamak için gereklidir.
Krabbe hastalığı hem lökodistrofi hem de lizozomal depo bozukluğu (LSD) olarak sınıflandırılır. Lizozomal depo bozuklukları, hücrenin lizozomundaki bir enzim eksikliğinden kaynaklanır. Lizozom, hücrenin geri dönüşüm merkezi olarak da bilinir ve Krabbe hastalığında olduğu gibi eksik veya eksik bir enzim olduğunda, vücut belirli maddeleri parçalayamaz ve bu maddeler hücre içinde birikir.
Krabbe hastalığından etkilenen bireyler, galaktoserebrosidaz (galaktosilseramid veya GALC olarak da adlandırılır) adı verilen spesifik bir lizozomal enzimi yeterince yapamazlar. Bu enzim eksikliği, galaktosilseramid (GALC) genindeki genetik bir değişiklikten kaynaklanmaktadır. Enzim eksikliğinin bir sonucu olarak, kompleks yağlı maddeler parçalanamaz ve depolama, özellikle globoid hücrelerin oluşumu ve sinirleri çevreleyen koruyucu kaplamanın/örtünün miyelin kaybı (demiyelinizasyon) gibi patolojik değişikliklere neden olur. Miyelin kaplı sinir lifleri, beynin beyaz maddesi olarak bilinir. Krabbe hastalığı, beyaz cevherin yanı sıra merkezi sinir sisteminin (MSS) geri kalanına (örneğin, beyin ve omurilik) ve MSS dışındaki sinirlerden oluşan periferik sinir sistemine yavaş yavaş zarar verir.
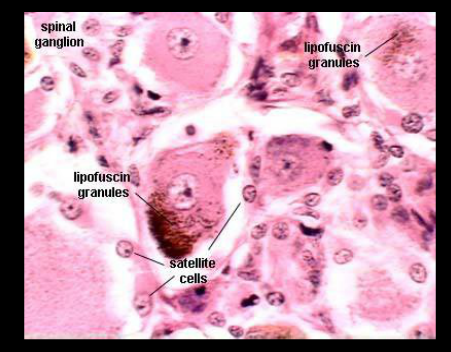
Nöronal Seroid Lipofuskinozis (NCL), benzer şikâyet ve bulgulara neden olan birkaç farklı genetik (ırsi) alt tipi olan nadir görülen nörodejeneratif hastalıklardır (Nörodejeneratif hastalık beyin ve sinir sisteminde ilerleyici bozulmaya neden olan hastalıklardır). Hastalığın farklı tiplerinin başlangıç zamanı ve ilerlemesi, genetik mutasyonun (bozukluğun) türüne ve diğer faktörlere bağlıdır. Bu bozuklukların hepsi sinir sistemini etkiler ve genellikle görme kaybı, epilepsi ve hareket ve bilişsel (zihinsel) yeteneklerde gerilemeye neden olur.
NCL’ye ne sebep olur?
Bugüne kadar, her biri farklı genetik mutasyonlara (bozukluk) bağlı olarak ortaya çıkan hastalığın 13 farklı tipi tanımlanmıştır. Her tipi, bozukluğa neden olan gen tarafından sınıflandırılır. NCL hastalığına neden olan ve bozukluk bulunan gene CLN denir ve her alt tipi için farklı bir sayı verilir. (NCL tip 1, NCL tip 2 NCL tip 3…)
Spinal Musküler Atrofi (SMA) hastalığı, omurilikteki hareket ettirici sinir hücrelerinde ilerleyici yıkım sonrası kas yıkımı ve güçsüzlüğü ile seyreden genetik bir hastalıktır.
SMA Hastalığının görülme sıklığı ne kadardır?
Ülkemizde SMA hastalığının görülme sıklığı ve taşıyıcılık oranları net olarak bilinmemekle birlikte, son yıllarda ülkemiz için yıllık yeni vaka sayısının 130-180 (ortalama: 150) arasında olduğu tahmin edilmektedir. T.C. Sağlık Bakanlığı Halk Sağlığı Genel Müdürlüğü Çocuk ve Ergen Sağlığı Daire Başkanlığı verilerine göre 2022 yılı itibariyle ülkemizde yaklaşık 3000 SMA hastasının izlendiği bilinmektedir. Hastaların gidişatı hastalığın tipine (SMA tip 1, SMA tip 2 vb.) bağlıdır. Hastalığın tipleri bulgularının başlama zamanı ve genetik sonuçlara göre belirlenmektedir.
SMA hastalığının sebebi nedir?
SMN1 isimli gende meydana gelen genetik mutasyonlar (bozukluk) nedeni ile SMA hastalığı görülmektedir.
Kaybolan Beyaz Madde Hastalığı (Vanishing White Matter) Nedir?
İnsan beyni gri madde ve beyaz maddeden oluşur. Gri madde sinir hücrelerini içerir. Beynin beyaz maddesi, sinir lifleri, miyelin, miyelin üreten oligodendrositler ve beyin dokusundaki düzenliliği koruyan astrositlerden oluşur. Miyelin, sinir liflerinin etrafına sarılmış yağlı bir kılıftır. Miyelin kılıfının iki işlevi; sinir uyarıları iletiminin yalıtımı ve hızlandırılmasıdır. Yalıtım, kısa devrelerin önlenmesi için önemlidir. Miyelin, sinir lifleri boyunca sinir uyarılarının yayılmasını hızlandırır.
Çocukluk çağındaki birçok nörolojik bozuklukta, beynin beyaz maddesinin bozukluğu söz konusu olabilir. Bunlara “beyaz cevher bozuklukları” (lökodistrofi) denir. Miyelin yeterli miktarda yapılmadığı için miyelin eksikliği olabilir. Bazı hastalıklarda miyelin oluşmuştur, ancak sonraki bir dönemde parçalanır ve kaybolur. Miyelinli lifler arasında artan miktarda su olabilir. Sinir lifleri de kaybolabilir. “Beyaz madde bozuklukları” birçok farklı hastalıkta ortaya çıkabilir ve beyin işlevlerinin sağlıklı yürütülmesini bozar. Kaybolan Beyaz Madde (VWM) hastalığı, bu yaygın beyaz cevher bozukluklarından (lökodistrofiler) biridir (OMIM numarası 603896).

")